
A Cleaning Validation Master Plan (CVMP) outlines the strategy, processes, and protocols to ensure equipment cleaning meets regulatory standards in pharmaceutical manufacturing, preventing contamination and ensuring product safety.

Table of Contents
1. Introduction
The Cleaning Validation Master Plan (CVMP) outlines the strategy, scope, responsibilities, and procedures for performing cleaning validation activities within the pharmaceutical manufacturing facility. This plan ensures that equipment used in the production of pharmaceutical products is cleaned effectively to prevent contamination, cross-contamination, and to maintain product quality in compliance with Good Manufacturing Practices (GMP) and regulatory requirements.
2. Purpose
The purpose of the CVMP is to provide a comprehensive and systematic approach to validation of cleaning activities within the facility. It ensures that:
Equipment cleaning processes consistently remove residues to predetermined and acceptable levels.
The risks of contamination and cross-contamination between different products are minimized.
Regulatory requirements are met for cleaning procedures.
3. Scope
This plan applies to all equipment, utensils, and cleaning processes involved in the production of drug products at the facility, including:
i. Manufacturing equipment (e.g., reactors, mixers, dryers)
ii. Analytical instruments in the Quality Control (QC) area
iii. Ancillary equipment (e.g., pipelines, filters, hoses)
iv. Cleaning agents used during the cleaning process
The CVMP covers cleaning validation for multi-product equipment, shared facilities, and utensils used in both drug substance and drug product manufacturing areas.
4. Objectives
The main objectives of this CVMP are:
i. To provide documented evidence that cleaning procedures are capable of consistently and effectively removing residues of active ingredients, excipients, cleaning agents, and microbial contaminants to acceptable levels.
ii. To define the acceptance criteria for cleaning validation.
iii. To ensure reproducibility of cleaning processes.
iv. To establish periodic revalidation protocols and assess the need for revalidation based on changes in process, product, or equipment.
5. Responsibilities
The responsibilities for executing and maintaining the cleaning validation program are as follows:
5.1 Quality Assurance (QA)
i. Approve cleaning validation protocols and reports.
ii. Ensure adherence to regulatory guidelines.
iii. Conduct internal audits of the cleaning validation process.
iv. Approve any changes to validated cleaning processes.
5.2 Production Department
i. Perform the cleaning procedures according to approved Standard Operating Procedures (SOPs).
ii. Document cleaning activities.
iii. Assist in cleaning validation execution.
5.3 Validation Department
i. Develop cleaning validation protocols and plans.
ii. Execute the validation studies, including sampling and testing.
iii. Analyze validation results and provide recommendations.
iv. Maintain the validation status.
5.4 Quality Control (QC)
i. Perform analytical testing on samples taken during cleaning validation.
ii. Validate analytical methods used for residue analysis.
5.5 Engineering
i. Ensure that equipment is designed and installed to facilitate effective cleaning.
ii. Provide technical support for equipment cleaning validation.
6. Validation Strategy
6.1 Types of Cleaning Validation
Initial Cleaning Validation: Performed when new equipment, products, or cleaning procedures are introduced.
Periodic Revalidation: Conducted as per validation schedule to confirm continued effectiveness of cleaning procedures.
Change Control Cleaning Validation: Triggered by changes in equipment, cleaning agents, processes, or products that may affect the cleaning procedure’s effectiveness.
6.2 Worst-Case Approach
A worst-case product approach will be used, selecting the product with the most difficult-to-clean properties (e.g., solubility, toxicity, potency) for validation studies. This ensures that if the worst-case product can be effectively cleaned, all other products can be cleaned as well.
6.3 Grouping of Equipment
Where feasible, similar types of equipment (e.g., tanks, mixers) will be grouped and representative equipment from each group will undergo validation.
7. Acceptance Criteria
Acceptance criteria for cleaning validations will be based on the following factors:
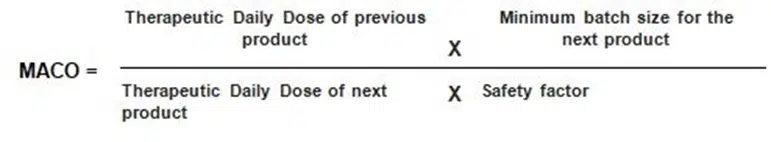
Maximum Allowable Carryover (MACO): The amount of residual product acceptable on equipment surfaces, calculated based on the therapeutic dose of the drug and safety limits.
Residual Limits for Cleaning Agents: Cleaning agent residues should be below the specified limits as per product safety data.
Microbial Limits: Total microbial count (TMC) should be within acceptable limits.
Visual Cleanliness: No visible residues should remain after cleaning.
Calculation of MACO:
8. Sampling Methods
i. Two primary methods will be used for residue detection:
ii. Swab Sampling: Used for hard-to-clean areas and surfaces with direct product contact.
iii. Rinse Sampling: Used to collect rinse solutions from equipment to detect residues in inaccessible areas.
iv. Both methods will be validated to ensure they provide accurate and reproducible results.
9. Analytical Methods
Analytical methods used for detecting residues will be validated for specificity, accuracy, precision, and limit of detection. These methods include:
i. High-Performance Liquid Chromatography (HPLC) for active ingredient residues.
ii. Total Organic Carbon (TOC) for organic residue detection.
iii. Spectrophotometric Methods for cleaning agents.
iv. Microbial Testing to assess microbial contamination.
10. Cleaning Validation Protocol
Each cleaning validation activity will follow a detailed protocol, outlining:
i. Purpose and scope of the validation.
ii. Equipment and cleaning procedures to be validated.
iii. Sampling locations and methods.
iv. Analytical methods and acceptance criteria.
v. Data recording and documentation requirements.
vi. Approval from QA and validation teams.
11. Revalidation
Revalidation will be triggered under the following conditions:
i. A change in product or process.
ii. Introduction of new equipment.
iii. A deviation from established cleaning procedures.
iv. After routine cleaning validations as per the CVMP schedule (e.g., every 2–3 years).
12. Change Control
Any changes to cleaning procedures, products, equipment, or facilities will be managed through a formal change control process. The need for revalidation will be evaluated as part of this process.
13. Training
Personnel involved in cleaning operations will receive appropriate training on cleaning procedures, validation protocols, and sampling techniques. Training records will be maintained as part of GMP documentation.
14. Documentation
All cleaning validation activities will be documented in:
i. Cleaning Validation Protocols
ii. Cleaning Validation Reports
iii. Cleaning Records (e.g., logbooks, batch records)
iv. Analytical Test Results
These records will be maintained according to GMP requirements and will be readily available for review during audits or regulatory inspections.
15. Conclusion
The Cleaning Validation Master Plan establishes the framework for ensuring the cleaning processes used in the facility consistently meet predetermined levels of cleanliness. By adhering to this plan, the company ensures compliance with GMP, maintains product quality, and minimizes the risk of cross-contamination.

Abdus Sobhan Salim is professional experienced pharmacist in pharmaceuticals, author and founder of pharmabossbd.com, the first Bangladeshi pharmaceutical blogger since 2019.


